
文章信息
● 原文: iMeta(IF 33.2, 中科院双一区Top)
● 英文题目:Soil-borne legacy facilitates the dissemination of antibiotic resistance genes in soil-plant continua
● 中文题目:土传微生物遗产驱动抗生素抗性基因在土壤-植物连续体中的传播
● 第一作者:肖祖飞、丁恺
● 通讯作者:李刚、Mui-Choo Jong(杨美珠)
● 主要单位:中国科学院城市环境研究所、宁波(北仑)中科海西产业技术创新中心、中国地质大学(北京)水资源与环境学院、清华大学深圳国际研究生院环境与生态研究院、杜伦大学(英国)
亮点
● 土传病害(如青枯病)显著加速ARGs在土壤-植物连续体中的传播与富集;
● 土传遗产可跨作物传播ARGs,影响后续作物安全;
● 构青枯菌侵染背景下,ARGs与MGEs、VFGs高度共现,存在向人类病原体转移的风险。
视频解读链接
Bilibili:https://www.bilibili.com/video/BV12cS7BJERx/
摘要
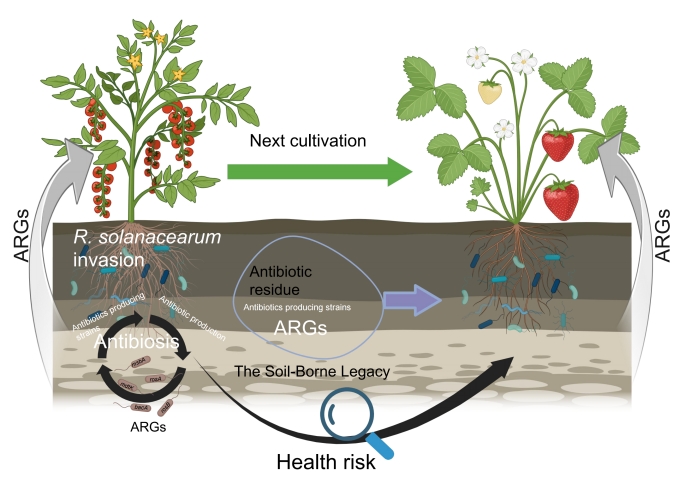
抗生素耐药性(AMR)借助复杂的微生物互作网络,在“土壤–植物”连续体中扩散。植物通过塑造根际与叶际微生物组维系自身健康;然而,农业生态系统中抗生素产生菌及其抗性基因所遗留的“土传遗产”,才是驱动AMR动态演替的“隐形之手”。本研究整合16S rRNA基因测序、宏基因组学与高通量qPCR技术,系统绘制了番茄–草莓双作物体系下土壤、根际、叶际及根内微生物组中抗生素抗性基因(ARGs)、可移动遗传元件(MGEs)与毒力因子基因(VFGs)的图谱。结果发现,细菌性青枯病的周期性暴发显著放大了抗性谱,多肽类抗性基因尤为显著,使根际成为ARG富集的“热区”。多重耐药的青枯菌(Ralstonia solanacearum)菌株作为核心ARG“蓄水池”,其染色体与巨型质粒共载多种抗性决定簇。综上,本研究首次揭示病原菌主导的植物微生物组重构可加速ARG在农业生态系统中的扩散,确立土传病害作为AMR放大的“关键倍增器”。
全文解读
引言
抗生素抗性基因(ARGs)从土壤环境扩散至作物、牲畜乃至人类生活圈,已成为全球公共卫生面临的重大挑战,也是“同一健康”(One Health)框架下的核心议题之一。尽管当前研究与政策多集中于人类医疗系统,但食物系统——特别是植物性农业生产,在抗生素耐药性(AMR)的出现与传播中所扮演的角色仍未得到充分重视。土壤作为ARGs和人类病原菌的重要储存库,其生态过程对AMR的扩散具有关键影响。随着农业集约化进程加快、耕地持续扩张以及“超级细菌”日益普遍,深入理解ARGs在土壤–植物连续体中的动态演变显得尤为紧迫。此外,对ARGs进行风险评估需综合考虑其类型、丰度、水平转移潜力以及宿主菌的致病性。当ARGs与毒力因子基因(VFGs)和可移动遗传元件(MGEs)共同出现时,可能进一步加剧微生物相关健康风险。
植物通过“求助”策略,主动招募根际微生物,塑造抑病型群落,形成所谓“土传遗产”。在抑病性土壤中,植物即使暴露于病原菌仍能维持健康生长,其机制主要包括:(1)有益微生物抢占生态位,与病原菌竞争养分和空间资源;(2)产生抗菌代谢物(如抗生素),抑制病原菌增殖、破坏细胞结构并削弱其毒力;(3)通过与植物根系建立共生关系,借助信号分子或代谢产物诱导植物系统抗性,激活宿主免疫反应以抑制病害发展。本研究重点关注上述机制(3)所引发的生态后果。土壤微生物间的拮抗作用通常由特定菌株在感知病原威胁后合成抗生素所触发。植物–病原菌的“军备竞赛”与有益菌–病原菌的“拮抗博弈”叠加,可能加速植物相关微生物组(土壤、根际、根内、叶际)中AMR的演化。农业中抗生素与生物防治剂的广泛使用,也进一步提高了植物病原菌的抗性水平。以青枯菌(Ralstonia solanacearum)为例,该菌天然耐受多种抗生素,可在含药培养基中被选择性富集。其存在还可能通过基因水平转移间接增强周边微生物群落(包括其他病原菌)获取ARGs的能力。然而,青枯菌所导致的抗性增强在多大程度上影响整个土壤–植物连续体中AMR的发展路径,目前尚不明确。
本研究以天然耐多药的青枯菌为模型,剖析一座具有长期青枯病发生历史的番茄温室,同时该病害还会影响到后续草莓作物的抗性组。我们提出假设:青枯菌施加的持续性选择压力会逐步富集特定ARGs,其影响跨越作物界限,从土壤–番茄连续体延伸至土壤–草莓连续体。为此,我们设定三大目标:(1)追踪青枯菌感染过程中抗性组与微生物组的动态变化;(2)评估ARGs是否可转移至后续土壤–草莓系统;(3)通过识别同时携带ARGs与MGEs的病原菌株,评估其潜在暴露风险(图1A)。通过结合高通量qPCR与宏基因组测序技术,我们系统绘制了土壤–植物连续体中ARGs、VFGs与MGEs的分布格局,从而揭示了青枯病在农业生态系统中对AMR传播的驱动作用。
结果
青枯菌入侵下细菌群落的变化
微生物群落分析表明,青枯菌入侵显著降低了细菌群落的丰富度与多样性,并引起根际、叶际和根内微生物群落结构的显著改变(图S1)。主要细菌类群(如γ-变形菌纲、α-变形菌纲、芽孢杆菌纲和放线菌纲)的相对丰度亦随感染状态发生显著变化(图S1、S2),表明青枯病严重扰乱了植物相关微生物群落的稳定性。群落多样性下降削弱了其生态缓冲能力,从而加剧了由病原菌驱动下的群落结构重塑。即使在未表现症状的植株中,青枯菌的存在仍可触发微妙的微生物拮抗作用,招募有益菌以抑制病害发展。此类微生物间的“军备竞赛”无意中促使产抗生素类微生物及其携带的ARGs得以富集(图S3、S4)。因此,病原菌介导的微生物组重构成为推动抗性组传播的关键生态机制。既往研究显示,健康植物微生物组中富含抗生素合成基因(如聚酮合酶和非核糖体肽合成酶),进一步印证了微生物间拮抗作用在塑造土传遗产中的核心地位。
抗性组在土壤–番茄连续体中的重塑
宏基因组分析共鉴定出1334种ARG亚型,分属29个类别。健康植株中ARG亚型的丰富度最高,其次分别为感染植株与死亡植株(表S1、S2)。抗性组主要由多药耐药类、杆菌肽类、多粘菌素类及β-内酰胺类抗性基因主导(图1B–D)。在所有组别中,根际均表现出最高的ARG、MGE和VFG的丰度与多样性,证实其为抗性传播的关键热点区域(图S5–S7)。研究结果显示,尽管有益菌能够抑制病害并维持植物健康,但其同时施加了强大的抗生素选择压力,促使ARG库不断富集。相反,病原菌感染则促进了MGE的动员以及临床相关ARG的累积,表明病原胁迫增强了水平基因转移(HGT)的潜力(图S11)。在土壤–植物连续体中,ARGs主要通过以下三条相互关联的途径进行传播:(1)经由气溶胶沉降或根系伤口被植物吸收;(2)根际中耐药菌占据优势,进而通过HGT将其ARGs转移至经由维管组织迁移的内生菌;(3)通过土壤动物(如蚯蚓)的摄食与排泄行为实现ARGs的再循环。这些过程在土壤细菌与植物内生菌之间建立起双向的基因流动网络,使ARGs得以进入食物链,最终对人类健康构成威胁。综上可知,在病原菌侵染的背景下,无论是抗病型还是感病型农业系统,均会通过不同的生态机制推动抗性组的传播与扩散,凸显出农业生态系统中ARGs管控的复杂性与重要性。
药敏试验进一步揭示了青枯菌对多粘菌素B、杆菌肽、氯霉素、青霉素、四环素及放线菌素D的耐药性存在显著差异(图S12)。分离得到的高致病性、多重耐药青枯菌株被确认为主要的ARG储存库。基因组分析表明,青枯菌的染色体和质粒上均编码对多粘菌素、杆菌肽、大环内酯、四环素、氟喹诺酮、香豆素、单环β-内酰胺、阿尔法霉素及氨基糖苷类药物的抗性基因(表S3)。基因型与表型的联合分析揭示了病原驱动选择如何放大土壤–植物连续体中具有临床意义的抗性特征,进而构成切实的食品安全风险。多模型整合分析进一步表明,青枯菌通过直接与间接两种生态机制驱动ARG的富集,而MGEs在其传播与动员过程中扮演关键媒介角色(图S13)。
土传遗产与跨作物ARG传播
对土壤–草莓连续体的分析表明,青枯病土传遗产的影响已超出番茄系统范围(图1E、F)。在曾发生青枯病的土壤中种植的草莓果实,其ARG的丰度与多样性均显著高于对照组(图S14A、S15,表S4)。HT-qPCR与宏基因组数据均证实,青枯病引发的土传遗产增强了ARG向植物组织迁移的潜力(图S16)。溯源分析进一步显示,草莓果实中66–89%的ARGs来源于健康番茄根际,表明存在跨物种的直接转移(图1G、S14B)。这一结果证实,土传遗产作为ARG在作物间迁移的储存库,扩大了青枯病的生态影响范围。值得注意的是,即使是健康番茄根际,仍是主要的ARG供体,说明即使未出现可见病害,抗性风险依然持续存在。这对食品安全构成直接威胁,因可食植物组织可能积累源自历史病原胁迫的ARGs。
尽管植物在表观上表现出对青枯菌的有效抑制,但病原驱动的选择性压力并未消失。相反,在健康状态阶段,系统内持续进行着强烈的“亚临床”微生物竞争。植物通过“呼救”招募有益菌,后者通过产生抗菌物质以抑制病原菌,但这一过程同时施加了强大的选择压力,富集了病原菌与有益菌中固有的抗性性状。在此条件下,病原菌、共生菌以及共生菌之间的HGT可能加速,促进ARG在根际MGEs上的整合与积累。此外,病原菌死亡后释放的细胞内容物、微生物群落结构的重塑、植物组织的加速分解以及HGT的促进,共同推动了ARG在土壤–植物连续体中的扩散。
土传遗产(亦称抑病性土壤记忆)在维持植物健康生长的同时,也驱动了ARG在土壤–草莓系统中的传播,其主要原因在于产抗菌物质细菌及相关ARGs的高丰度存在。这类细菌包括放线菌、芽孢杆菌、假单胞菌和溶杆菌等,它们携带多种生物合成基因簇与ARGs,与链霉菌共同参与抗生素合成和病害抑制过程,同时也成为ARG的重要储存库。ARGs、MGEs与青枯菌丰度之间的正相关关系表明,青枯病是ARG在土壤–植物连续体中传播的关键载体,对食品安全与人类健康构成严重威胁,亟需加强对该类路径的系统监测。
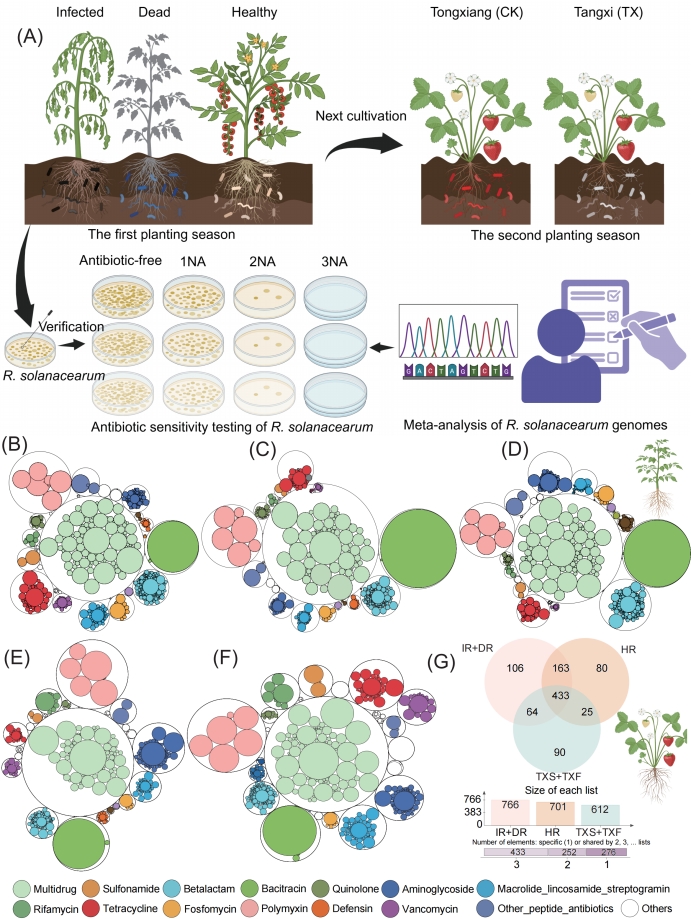
图1. 土传遗产促进ARGs在土壤-植物连续体中的传播
(A)实验设计流程图。本研究共包括四项实验:两次田间调查与两次验证性试验。首次田间调查旨在表征染病、健康及濒死番茄植株对应的土壤-番茄连续体中的抗性组与微生物组(涵盖细菌群落、ARGs、毒力因子基因VFGs、可移动遗传元件MGEs及相关代谢通路)。第二次田间调查于番茄收获后,评估了种植于同一地块的草莓植株中的抗性组。第一项验证性试验通过药敏试验评估了青枯菌分离株的抗生素耐药性;第二项验证性试验则通过基因组分析鉴定了该菌分离株中的ARGs,并评估了其耐药性。(B–D) 不同植株健康状态下,土壤-番茄连续体中抗生素抗性组的分布与组成。抗性组按类别展示,内圈代表各ARG亚型,外圈表示其所属的ARG类别;圆圈大小反映对应组别(健康B、感染C、死亡D)中经标准化处理的ARG相对丰度。(E–F) 不同生态位下,土壤-草莓连续体中ARGs的分布、组成及来源解析。抗性组构成以相同方式呈现,内圈为亚型,外圈为类别,圆圈大小分别对应对照组(CK,E)和处理组(TX,F)中标准化后的ARG丰度。(G)溯源分析预测土壤-草莓连续体中ARGs的来源。
缩写说明:NA,营养琼脂培养基;HR,健康番茄根际土壤;IR,感染番茄根际土壤;DR,死亡番茄根际土壤;CK,来自桐乡、无青枯病史的土壤-草莓连续体;TX,来自塘溪、有青枯病史的土壤-草莓连续体;TXF,TX组的草莓果实;TXS,TX组的草莓根际土壤。
抗性基因的动员及其公共卫生风险
基于宏基因组组装基因组(MAG)的分析表明,ARG在致病菌(如大肠杆菌、肺炎克雷伯菌)和有益菌(如芽孢杆菌、假单胞菌)中均频繁与MGE及VFG发生共定位(图2A,表S5–S10)。这些基因在相同contig上的共存,表明其具备较高的动员潜力,能够促进跨不同微生物类群的HGT。在contig水平上,我们进一步识别出多药耐药、β-内酰胺类和氨基糖苷类抗性基因与转座酶、整合酶以及质粒复制基因在物理位置上紧密连锁,为ARG的移动性提供了直接证据(图2B–D,表S11–S14)。此类共定位现象,尤其在病原菌和植物共生菌中普遍存在,提示农业土壤——特别是受青枯病影响的土壤,可能作为HGT的储存库与传播通道,进而潜在地促进具有临床意义的耐药菌株的出现。
上述发现凸显了土壤微生物组所具有的“双刃剑”特性:一方面,用于病害防治的生防菌本身可能成为ARG的储存库;另一方面,病原菌则可能获取这些抗性基因,进一步增强其在临床环境中的适应性与威胁。一个尤为值得关注的发现是多肽类抗性基因的富集,这类基因会削弱多粘菌素和万古霉素等“最后防线”抗生素的疗效。非核糖体抗菌肽(AMP)曾因毒性较低和作用特异性而被视为低风险抗菌药物,但其有效性如今也因抗性基因的扩散而受到威胁。尽管多种病原菌和土壤益生菌天然产生AMP以在生态位竞争中取得优势,但ARG的传播正在削弱这类化合物在生态和临床场景中的效力。更令人担忧的是,本研究在青枯病影响的土壤及其所种植的草莓果实中均检测到万古霉素抗性基因的存在,进一步强调农业土壤作为生态与临床耐药性交汇的关键界面,对食品安全与公共健康构成直接且现实的威胁。
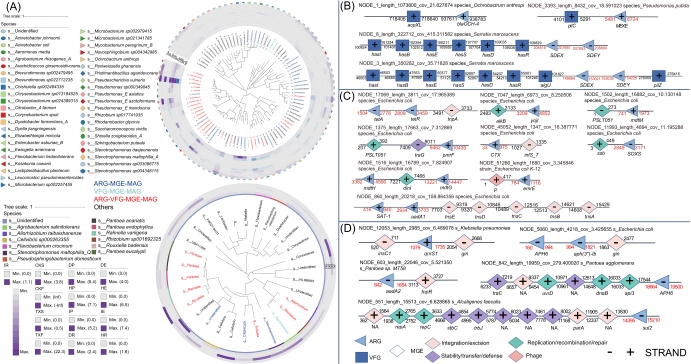
图2. 土壤-番茄与土壤-草莓连续体中携带ARGs、MGEs和VFGs的微生物宿主
(A)土壤-番茄(左上)与土壤-草莓(左下)连续体中MAGs的系统发育树,按携带ARG-MGE、VFG-MGE、ARG-VFG-MGE的菌株及其他类别着色。(B)土壤-番茄连续体中携带ARG-MGE簇的代表性重叠群及其推测宿主(NCBI最佳匹配)。(C)土壤-番茄连续体中携带ARG-VFG簇的代表性重叠群及其推测宿主(大肠杆菌)。(D)土壤-草莓连续体中携带ARG-MGE簇的代表性重叠群及其推测宿主。数字表示基因的起始位置;红色与黑色分别对应ARGs及其他基因。缩写说明:番茄连续体——HR,健康植株根际;HE,健康植株根内;HP,健康植株叶际;IR,感病植株根际;IE,感病植株根内;IP,感病植株叶际;DR,死亡植株根际;DE,死亡植株根内;DP,死亡植株叶际;InR,发病前根际。草莓连续体——CKF,桐乡无病果实;CKS,桐乡无病根际土;TXF,塘栖果实;TXS,塘栖有青枯病史的根际土。
综合视角
总体而言,本研究结果表明,即使在无症状阶段,青枯菌仍能持续促进ARG在土壤–番茄连续体中的存续与扩散。青枯病所引发的土传遗产可跨越单一作物周期,促进ARG向后续作物及其可食组织迁移。ARG、MGE和VFG在多种微生物宿主中的频繁共现,揭示了ARG在农业环境中的多重传播路径,凸显了将植物病理学、微生物生态学与“同一健康”框架整合纳入AMR治理体系的紧迫性。本研究进一步强调,病原胁迫、微生物互作及HGT共同塑造了农业生态系统中抗性组的动态演变。此外,未来在开发微生物肥料和生物防治制剂时,应建立双重评估标准:在考察其病害防控效果的同时,也需最大限度地降低其所携带的ARG负荷,避免无意中加剧抗生素抗性这一公共卫生挑战。
然而,本研究仍存在若干局限性。短读长宏基因组测序技术对质粒等MGE结构的解析能力有限,影响了ARG和MGE向其宿主基因组精确归属的可靠性。土壤pH、有机质含量及农药使用等环境因素未作为独立变量加以控制,尽管实验在标准化管理下进行,但这些因子仍可能影响ARG的分布格局。作为一项横断面研究,本研究未能捕捉ARG的长期动态变化或微生物群落的演替过程。此外,宏基因组测序与高通量qPCR方法均受到植物宿主DNA污染的干扰,限制了其在绝对定量方面的准确性。尽管宏基因组技术能够提供广谱的ARG谱图,而HT-qPCR对稀有但高风险ARG的检测更为灵敏,方法学差异仍可能导致部分结果存在轻微不一致。
基于上述局限,未来研究应重点关注以下方向:(1)采用长读长测序或Hi-C分箱等技术,精确解析ARG–MGE–宿主的基因组背景;(2)结合宏转录组学与功能性实验,揭示病原胁迫下ARG的活跃表达及其介导的微生物互作机制;(3)开展长期定位观测与时间序列分析,追踪不同季节及农业管理措施下ARG的动态演变规律;(4)设计受控实验,系统阐明有机质、农药残留等关键环境因子对抗性组传播行为的影响。
资讯来源:微信公众号”iMeta“推文《iMeta | 中科院朱永官院士李刚组-解析土传遗产在抗生素抗性基因传播中的作用》
阅读推文
阅读原文