
近年来,全氟化合物(PFAS)作为现代工业的 “明星材料”,广泛应用于氟材料、锂电池和半导体等领域。然而,其极强的环境持久性与生物毒性,使其成为全球关注的“永久性污染物”。PFAS分子中牢固的C-F键(键能高达485 kJ/mol)是其难以降解的“罪魁祸首”,如何高效断裂C-F键、实现PFAS彻底降解,成为环境科学领域的前沿挑战。
西湖大学张岩岩实验室在《Nature Water》发表的研究成果,首次揭示了水合电子降解PFAS的 “电子转移限速机理”,为破解这一难题提供了全新思路。该研究通过实验与理论计算结合,系统剖析了41种PFAS的降解行为,发现水合电子降解PFAS的决速步骤并非传统认知中的C-F键断裂,而是电子从水合电子向PFAS分子的转移过程。这一发现颠覆了既往研究对PFAS降解机制的理解。
关键发现:电子转移主导降解速率,官能团调控反应路径
研究团队通过 UV/亚硫酸盐体系生成水合电子,发现不同PFAS的降解速率常数差异达4个数量级。理论计算表明,电子转移的活化自由能(ΔGET)与降解速率常数(kdeG)呈现高度负相关(R²=0.744,p<0.01),证实电子转移是反应的限速步骤。例如,含C=C双键或C-Cl键的PFAS,因电子转移能垒低,降解速率比含短链(CF2) n≤3的化合物快100倍以上。
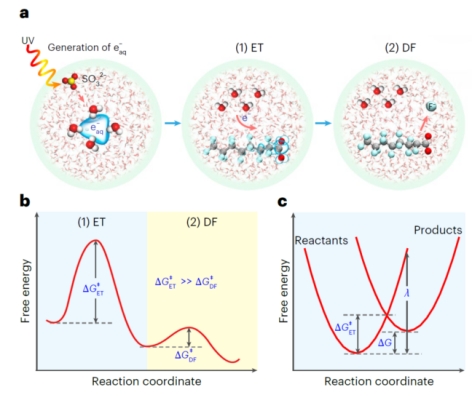
图1. 水合电子降解PFAS的反应过程:电子转移(electron-transfer,ET)和C−F键断裂(defluorination,DF)
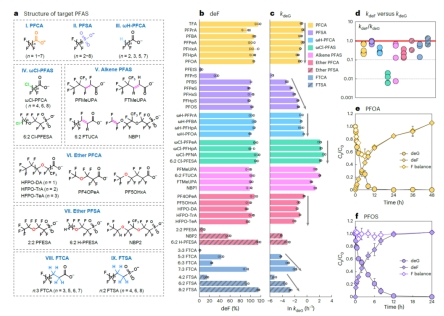
图2. UV/亚硫酸盐降解PFAS的脱氟效率(deF)、降解速率常数(kdeG)和典型动力学趋势
进一步研究发现,官能团通过影响电子转移效率调控降解路径:
促进电子转移的官能团:C=C、C-Cl、CF2COO−和长链 (CF2) n≥6,可显著降低电子转移能垒。如PFOS(含长链CF2)的中间产物电子转移能垒低,实现“同步脱氟”,氟离子释放与分子降解同步完成。
抑制电子转移的官能团:C-H、-O-、短链(CF2) n≤3和SO3−,会阻碍电子转移。以PFOA为例,其CF2COO−邻近的α-CF2位点优先脱氟,但生成的中间产物电子转移能垒高,导致“脱氟滞后”,氟离子释放显著延迟。

图3. 水合电子降解PFAS的电子转移限速机理:提出与验证的逻辑框架

图4. (H2O)4-eaq-分子簇模拟水合电子及合理性验证
双重脱氟路径:从分子机制到污染治理的桥梁
该研究首次提出PFAS降解的两种典型路径:
同步脱氟型:长链PFSA、FTSA等化合物,电子转移优先发生在(CF2) n≥6区域,中间产物能垒低,迅速连续脱氟,氟质量平衡始终维持在100%。
滞后脱氟型:PFOA及其衍生物,电子转移集中于CF2COO−头基,生成的中间产物与水合电子反应活性差,导致氟离子释放滞后,实验中可检测到6种高能量中间产物累积。
这种路径分化机制为复杂PFAS混合物的治理提供了理论依据。例如,针对工业废水中同时存在的PFOA和PFOS,可通过调控反应条件(如pH、氧化剂浓度),优化电子转移效率,实现高效脱氟。
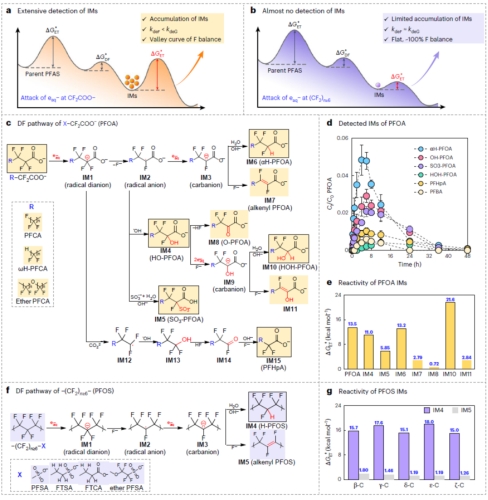
图7. 电子转移限速机理解析水合电子降解PFAS的两种典型脱氟模式
从基础理论到应用实践:绿色替代物设计的 “路线图”
基于官能团调控机制,研究团队建立了PFAS电子转移促进能力的官能团序列:C=C > C-Cl > CF2COO− > (CF2) n≥6 > C-H ≈ (CH2)n > SO3− > (CF2) n≤3 ≈ -O-。这一序列为设计易降解的PFAS绿色替代物提供了明确指导:即在材料设计中引入C=C双键或C-Cl基团,可在保持材料性能的同时,显著提升其可降解性;避免使用以SO3−和短链(CF2) n≤3为骨架的结构,减少环境持久性风险。
未来挑战:从实验室到实际环境的跨越
尽管该研究在机理层面取得重大突破,但实际应用仍面临挑战:
反应体系拓展:目前机理验证限于UV/亚硫酸盐体系,在UV/吲哚、UV/碘等其他水合电子体系中的普适性需进一步验证。
环境基质影响:实验室理想条件(无氧、纯水、pH=12)与实际水体(含溶解氧、腐殖质、pH=7)差异显著,需研究基质组分对电子转移效率的干扰机制。
张岩岩研究员表示:“破解PFAS污染难题,需要从‘末端治理’转向‘源头防控’。我们的研究不仅揭示了降解机理,更希望为绿色化学品设计提供‘分子工具箱’,推动产业向环境友好型转型。”
该研究得到国家自然科学基金委、浙江省自然科学基金委和西湖大学未来产业研究中心的支持,为全球PFAS污染治理提供了中国学者的智慧方案,也为新污染物环境行为研究开辟了“理论—实验—应用”的一体化研究范式。
资讯来源:微信公众号”环境生态网“推文《西湖大学张岩岩研究员团队破解PFAS降解难题:揭示C-F键断裂新机制,为新污染物治理开辟路径》
阅读推文
阅读原文