
研究论文
● 期刊: iMeta (IF 33.2, 中科院双一区Top)
● 2025年8月3日,中山大学谢秀琴、陈保卫、栾天罡和暨南大学附属广东省第二人民医院程伟彬等在iMeta在线发表了题为“Functional Metagenomics Reveals Novel Antibiotic Resistomes in Polar Soils”的文章。
● 本研究使用功能宏基因组学的方法,识别了南北极土壤样品中的抗生素新型抗性基因,系统评估了其移动性、宿主致病性和传播潜力。这项研究从以前被忽视的极地地区的抗生素新型抗性基因的视角,促进我们对环境抗性组的理解。
● 第一作者:谢秀琴、程伟彬
● 通讯作者:陈保卫(chenbw5@mail.sysu.edu.cn)
● 合作作者:李昭宏、何荣、原珂、张庆华、杨瑞强、明荔莉、余珂、栾天罡
● 主要单位:中山大学海洋科学学院、暨南大学附属广东省第二人民医院、澳门城市大学健康科学学院、广东省化学与精细化工实验室揭阳中心、中国科学院生态环境科学研究中心、珠海拱北海关技术中心、北京大学深圳研究生院、中山大学生命科学学院
亮点
● 使用功能宏基因组学方法,在极地土壤中发现了对临床抗生素具有抗性的新型抗性基因;
● 与已知同类抗性基因相比,极地抗生素新型抗性基因具有移动性差和宿主致病风险低的特点;
● 极地抗生素新型抗性基因可以作为合适的标记物,有效区分受不同程度人类影响的环境抗生素抗性组。
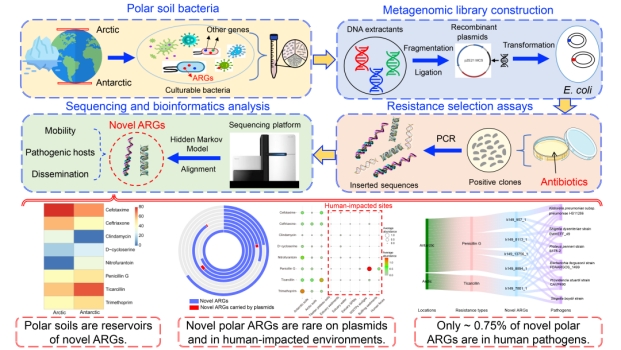
引言
抗生素耐药性是一个全球性的健康问题,具有抗生素耐药性的细菌病原体每年可导致数十万人死亡。在永久冻土环境中发现了多种携带抗生素抗性基因(Antibiotic resistance genes,ARGs)的潜在人类病原体(如Serratia liquefaciens和Yersinia enterocolitica)。据估算,因全球变暖,每年约有4.0×1021个微生物细胞从冰冻封存环境中释放出来,这些冰川微生物可能入侵水生和陆地生态系统。冰封微生物携带的ARGs也将从极地区域释放。研究证实,极地ARGs可在微生物间发生水平基因转移,最终可能导致临床问题。
极地环境是重要的ARGs储库。我们前期研究发现,极地土壤中的可培养细菌群落对多种临床抗生素具有耐药性,但其基因组中缺乏相应的已知ARGs。这表明极地抗生素抗性组仍未被充分认识。功能宏基因组学(Functional metagenomics)通过在替代宿主中异源表达宏基因组DNA片段,并结合功能筛选、高通量测序和生物信息分析,从宏基因组序列中发现功能尚不明确的基因。该方法不依赖于与已知基因的序列相似性,可鉴定功能验证的ARGs,能深入探索抗性组。凭借其相对于传统分子生物学检测和基于序列的宏基因组学分析的优势(如高通量、不需要已知基因的功能注释),功能宏基因组学已成功从多种环境样品中识别抗生素新型抗性基因。全面表征极地抗生素新型抗性基因对其传播管控和健康风险防控至关重要,例如将抗生素新型抗性基因纳入基因诊断和靶向监测体系。本研究综合探究了北极与南极土壤中的抗生素新型抗性基因,系统评估其移动性、传播潜力及潜在宿主致病性。
研究结果
极地土壤中经过功能验证的多种抗生素新型抗性基因的鉴定
功能宏基因组方法能对抗性组进行深入分析,不依赖已知ARGs,识别赋予抗生素耐药性的DNA片段。我们使用鸟枪法克隆策略,将来自极地土壤可培养细菌的DNA片段(约1.5 kb)克隆至大肠杆菌(Escherichia coli),构建了8个宏基因组文库(南极:0.05–2.1 Gb;北极:0.07–0.29 Gb)(表S1),然后针对9大类23种抗生素进行耐药性筛选(方法见补充材料)。在检测的23种用于筛选的抗生素中,极地土壤宏基因组对其中的8种表现出抗性(表S2)。随后,对具有抗生素耐药性的DNA片段进行测序、组装与注释(方法见补充材料)。
在345,833个开放阅读框(open reading frames, ORFs)中,注释出18,657个ARGs。其中329个(北极)与342个(南极)ORFs达到已报道的抗生素新型抗性基因的筛选标准(图1A),在极地ARGs总量中占比较低(图S1A)。值得关注的是:约20.0%的极地抗生素新型抗性基因与其最相近的NCBI同源基因相似度小于80.0%(图1B)。大约70.0%的抗生素新型抗性基因能够对β-内酰胺类抗生素(例如头孢噻肟和替卡西林)产生抗性,其次是叶酸合成抑制剂(约14.2%)、D-环丝氨酸(约6.4%)、硝基呋喃(约4.9%)和克林霉素(约4.8%)。北极和南极土壤中抗生素新型抗性基因的组成存在显著差异(two-sided Fisher’s exact test,p < 0.05)(图S1B)。
极地抗生素新型抗性基因具有低移动性
通过与NCBI质粒数据库进行比对,评估了极地抗生素新型抗性基因与已知ARGs(来源:CARD数据库)在细菌宿主间的水平转移潜力。超过75.0%的已知ARGs(β-内酰胺类/呋喃妥因/甲氧苄啶相关)被质粒携带,而极地抗生素新型抗性基因极少被质粒所携带(图1C)。仅发现7个极地抗生素新型抗性基因被质粒携带,其中与青霉素G相关的有4个,替卡西林相关的为1个,克林霉素相关的为2个。值得注意的是:在超过690种质粒类型上发现青霉素G(β-内酰胺类)相关的新型抗性基因(图1D)。对于β-内酰胺抗性基因,编码A类β-内酰胺酶的已知ARGs在质粒上的出现频率显著高于极地抗生素新型抗性基因(p < 0.05)(图S2)。
极地抗生素新型抗性基因在人类致病菌基因组中的出现率低
通过比对人类致病菌数据库,我们分析了极地抗生素新型抗性基因与前述已知ARGs的出现模式。已知ARGs广泛存在于致病菌宿主中,覆盖了所有极地抗生素新型抗性基因的类别,且在每个类别中占比均超过25.0%(图1E)。相比之下,仅有0.75%的极地抗生素新型抗性基因能在致病菌基因组中发现,且这些基因均表达对β-内酰胺类抗生素的耐药性。值得注意的是,有4种抗生素新型抗性基因分别被6种致病菌携带,包括肺炎克雷伯菌(Klebsiella pneumoniae)、痢疾志贺菌(Shigella dysenteriae)、鲍氏志贺菌(Shigella boydii)、彭氏变形杆菌(Proteus penneri)、弗格森埃希菌(Escherichia fergusonii)以及斯氏普罗威登斯菌(Providencia stuartii)(图1F)。这些与致病菌相关的抗生素新型抗性基因均被质粒携带(图1D,F)。针对β-内酰胺抗性基因,新型与已知抗性基因在致病菌宿主中的出现频率及宿主组成均在统计学上存在显著差异(Kruskal-Wallis test,p < 0.05)(图S3A,B)。
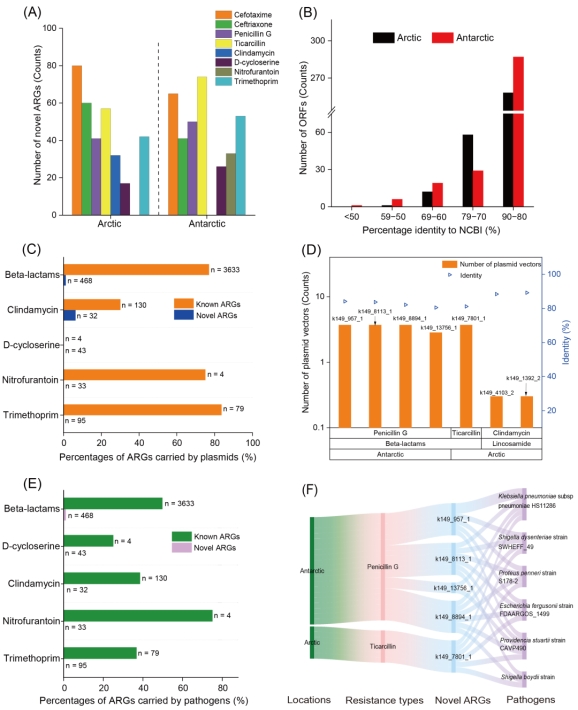
图1. 对临床抗生素表达耐药性的极地抗生素新型抗性基因的独特特征
(A)按抗生素类型分类的北极和南极土壤中抗生素新型抗性基因的数量。(B)被认定为极地抗生素新型抗性基因的ORFs与其在NCBI蛋白质数据库中的最佳匹配基因之间的氨基酸相似性(黑色代表北极(329);红色代表南极(342))。(C)质粒携带的新型和已知ARGs占总ARGs的百分比。‘n’代表用于比对分析的ARGs序列总数。(D)质粒携带的极地抗生素新型抗性基因的总结,包括携带抗生素新型抗性基因的质粒载体数、抗生素种类和与已知ARGs的相似度。其中,对质粒载体的数量进行了对数转换。(E)致病菌携带的ARGs占总ARGs的相对百分比。‘n’代表用于比对分析的ARGs的序列总数。(F)在北极和南极土壤中鉴定出的抗生素新型抗性基因的人类致病宿主预测。
极地抗生素新型抗性基因在人类影响环境中的有限传播
我们分析了不同环境宏基因组数据中的极地抗生素新型抗性基因(表S3),这些环境涵盖了由抗生素使用导致的典型抗生素抗性基因污染源(例如废水处理厂和水产养殖场),以及受人类活动影响较小的相对原始地区(例如西藏)。在九个宏基因组数据集中共检测到138种极地抗生素新型抗性基因,其总丰度从未检出(ND)(D1河口沉积物)至每百万测序读数中11.8条ARG-like(S2南极土壤)不等(图2A),其中β-内酰胺类(65.2%)与甲氧苄啶类相关(15.2%)抗性基因的丰度最高。极地抗生素新型抗性基因在原始环境中的检出率显著高于人类影响环境(图2A)。西藏土壤中含有60种极地抗生素新型抗性基因,而7个人类影响样品中仅检出29种极地抗生素新型抗性基因。通过已知同类ARGs的对比分析发现,原始环境中抗生素新型抗性基因数量显著超过已知ARGs(p < 0.05;图2B),而在人类影响环境中则呈现相反趋势(图2C)。主成分分析(PCA)显示,基于抗生素新型抗性基因的组成,原始环境和人类影响环境之间存在统计学上显著的差异(ANOSIM,p < 0.05;图2D)。
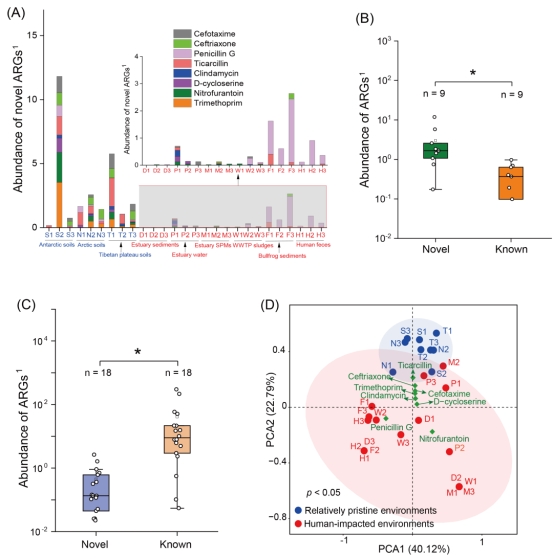
图2. 极地土壤中检出的抗生素新型抗性基因在自然环境中普遍存在
(A)受不同程度的人类影响环境中极地抗生素新型抗性基因的丰度,S1、S2等分别代表来自不同环境的采样点。(B-C)比较相对原始(B)和受人类影响(C)环境中新型和已知ARGs的总丰度,其中“*”表示通过Kruskal-Wallis test得出的具有统计学意义的差异(p < 0.05),“n”表示用于此次分析的测序数据集数量。“1”表示ARGs丰度的单位,为每百万测序读数中一个ARGs片段。(D)使用主成分分析(PCA)方法根据极地抗生素新型抗性基因的丰度对采样点进行聚类分析。
结果讨论
未受人为干扰的原始环境生态位被广泛认为是主要的ARGs储库。然而,环境抗性组的功能多样性常被低估,因为已鉴定的土壤ARGs与公共数据库收录的ARGs存在极大的差异。例如在中国云南、四川和西藏的原始区域(无抗生素污染)土壤样本中,曾鉴定出24种四环素新型抗性基因。有研究在无合成抗生素药物暴露史的森林土壤中也发现了磺胺类新型抗性基因。本研究证实了极地土壤中存在与临床抗生素相关的经过功能验证的抗生素新型抗性基因。更重要的是,这些抗生素新型抗性基因的发现揭示了传统宏基因组学方法的局限性,即仅依赖公共数据库无法全面解析环境抗性组。本研究用于耐药性筛选的抗生素中,青霉素G和D-环丝氨酸可能为微生物天然产物,其余则为合成或半合成抗生素。值得注意的是,在极地样本中同样发现了对合成/半合成抗生素具有抗性的新型基因。这表明自然环境中的本土微生物基因组是抗性发展和传播的基因储库,甚至对合成抗菌剂具有抗性。
本研究证明,极地土壤中发现的抗生素新型抗性基因在细菌宿主间通过质粒进行水平转移能力有限。该特性在其他环境的抗生素新型抗性基因的研究中被报道,例如美国农业与草地土壤中鉴定到的抗生素新型抗性基因未与移动遗传元件共现。另外,前抗生素时代的微生物区系(如肠杆菌科)也鲜有ARGs被可移动基因元件携带。此外,原始环境中已知ARGs也同样呈现低移动性,西藏生态系统中质粒携带ARGs的稀缺性也证明了这一点。与此对比显著的是,人类影响环境中质粒携带的ARGs丰度显著更高。这些发现意味着,选择压力缺失有助于形成原始环境抗性组的关键特征:即极低的移动性。
环境ARGs储库被认为是人类病原体抗生素耐药性的潜在源头。本研究发现,仅极少量极地抗生素新型抗性基因(约0.75%)可能存在于人类致病菌中,表明这些抗生素新型抗性基因当前的健康风险有限。已有研究报道表明,在2895个土壤ARGs中仅有1个与病原体相关(基因相似度为100%)。值得警惕的是:人类致病菌基因组中抗生素新型抗性基因的检出表明环境储库与人类致病菌间的基因交换壁垒可能被突破。有研究发现人类致病菌中的ARGs较非致病菌表现出更强的水平基因转移潜力,此结果与本研究一致,这种特性可能加速ARGs在人类致病菌间的传播。致病菌获得抗生素新型抗性基因能增强其抗生素耐药性,可能增加不可预见的临床负担。
极地土壤中发现的抗生素新型抗性基因在人类影响环境中检出率极低,表明其传播能力有限。与此形成鲜明对比的是,已知同类ARGs在人为干扰环境中持续富集,这与前期研究结果高度一致。有趣的是,尽管与极地相距遥远,极地抗生素新型抗性基因在西藏土壤中的检出率与丰度也很高。综上所述:极地抗生素新型抗性基因有助于区分相对原始环境与人类影响环境中的抗性组。本研究表明,在没有人类影响的原始环境中,极地抗生素新型抗性基因可以作为ARGs组成的有效标记物。
结论
本研究证明极地环境是抗生素新型抗性基因的重要储存库。在极地土壤中发现的抗生素新型抗性基因表现出有限的水平转移能力性、传播潜力和致病风险。本研究还强调,使用宏基因组学方法研究已知ARGs可能忽略了大量尚未被表征的ARGs,从而低估了自然环境中抗生素抗性组的多样性。
研究方法
详细的实验材料和步骤,包括样品采集和处理,以及统计分析方法在补充信息中进行了详细描述。
作者简介

资讯来源:微信公众号”iMeta“推文《iMeta | 中山大学陈保卫组-解析极地土壤中抗生素新型抗性基因》
阅读推文
阅读原文